Ansys Fluent可以通过求解描述对流、扩散和每个组分组分反应源的守恒方程来模拟化学组分的混合和传输。可以模拟多个同时发生的化学反应,反应发生在流体相(体积反应)、壁面或颗粒表面,以及多孔区域。本节描述了有无反应的组分传输建模能力。
请注意,您可能还希望使用混合分数方法(适用于非预混系统,参见非预混燃烧(第295页))、反应进度变量方法(适用于预混系统,参见预混燃烧(第332页))、部分预混方法(参见部分预混燃烧(第343页))或成分PDF传输方法(参见成分PDF传输(第276页))来模拟湍流反应火焰。有关使用有限速率化学的模拟多相组分传输,请参见多相流(第595页)。
信息分为以下部分:
- 7.1.1. 体积反应
- 7.1.2. 壁面表面反应和化学气相沉积
- 7.1.3. 颗粒反应
- 7.1.4. 电化学反应
- 7.1.5. 反应通道模型
- 7.1.6. 反应器网络模型
有关在Ansys Fluent中使用这些模型的更多信息,请参阅用户指南中的“模拟组分传输与有限速率化学”。
7.1.1 体积反应
本节介绍了与体积反应相关的组分传输和有限速率化学的理论信息。这些信息分为以下几个部分:
7.1.1.1 组分传输方程
7.1.1.2 反应建模的广义有限速率公式
7.1.1.3 双温度模型下的有限速率化学
有关使用与体积反应相关的组分传输和有限速率化学的更多信息,请参阅用户指南中的体积反应部分。
7.1.1.1 组分传输方程
当您选择求解化学组分的守恒方程时,Ansys Fluent 通过求解第 组分的对流-扩散方程来预测每个组分的局部质量分数 。该守恒方程采用以下一般形式:
混合物密度
扩散组分的速度
组分 通过化学反应产生的净生成速率(本节后面描述)
通过分散相添加以及任何用户定义的源项的创建速率。这种形式的方程将针对 个组分进行求解,其中 是系统中存在的流体相化学组分的总数。由于组分的质量分数必须总和为1,因此第 个质量分数由1减去求解的 个质量分数的总和来确定。为了最小化数值误差,第 个组分应选择为具有总体最大质量分数的组分,例如当氧化剂为空气时选择 。
7.1.1.1.1 层流中的质量扩散
在公式 7.1(第238页)中, 是组分 的扩散通量,这是由于浓度和温度梯度引起的。默认情况下,Ansys Fluent 使用稀释近似(也称为菲克定律)来模拟由于浓度梯度引起的质量扩散,在此近似下,扩散通量可以写为
其中
混合物中组分 的质量扩散系数
组分 的质量分数
热(Soret)扩散系数(参见热扩散系数 (p. 242))
温度
对于某些层流流动,稀薄近似可能不适用,需要完全多组分扩散。在这种情况下,可以求解 Maxwell-Stefan 方程;详细信息请参见用户指南中的完全多组分扩散 (p. 239)。
方程 7.2 (p. 238) 严格有效当混合物组成不变,或者当 与组成无关时。这是一个在稀薄混合物中的可接受近似,当 ,对于所有 除了载气。Ansys Fluent 还可以通过将此类混合物视为多组分系统来计算层流流动中的非稀薄混合物的传输。在 Ansys Fluent 中, 可以通过多种方式指定,包括通过指定 ,组分 在组分 中的二元质量扩散系数。然而, 并不直接使用;相反,混合物中的扩散系数 计算为 [299] (p. 1074):
其中, 和 分别表示组分 的摩尔分数和质量分数, 表示孔隙率。你可以根据 Fluent 用户指南中关于质量扩散系数输入的描述,为每个化学组分输入 或 。
7.1.1.1.2 湍流中的质量扩散
在湍流中,Ansys Fluent 采用以下形式计算质量扩散:
其中 表示湍流粘度,而 则是通过以下公式计算的湍流Schmidt数:
其中, 表示湍流扩散系数。 的默认值为 0.7。需要注意的是,湍流扩散通常会主导层流扩散,因此在湍流流动中通常不需要详细指定层流扩散特性。
在湍流流动中,总扩散是层流扩散 (由方程 7.3 (第 239 页) 给出)与湍流扩散(由方程 7.5 (第 239 页) 给出)之和。
7.1.1.1.3 完全多组分扩散
当分子传输过程的细节变得重要时(例如,在扩散主导的层流流动中),对组分传输和能量方程中的化学组分扩散进行仔细处理是重要的。作为层流扩散模型之一,Ansys Fluent 具备模拟完全多组分组分传输的能力。
7.1.1.1.3.1 一般理论
对于多组分系统,通常无法推导出仅包含单一组分梯度的扩散通量关系(如第 238 页的层流中的质量扩散所述)。在这里,将使用麦克斯韦-斯蒂芬方程来获得扩散质量通量。这将导致广义菲克定律扩散系数的定义 [649] (第 1094 页)。与计算多组分扩散系数相比,这种方法更受欢迎,因为它们的评估需要计算大小为 的 个协因子行列式,以及一个大小为 的行列式 [636] (第 1093 页),其中 是化学组分的数量。
7.1.1.1.3.2 麦克斯韦-斯蒂芬方程
根据 Merk [436] (第 1082 页),麦克斯韦-斯蒂芬方程可以写为
其中,
表示摩尔分数,
表示扩散速度,
表示组分 在组分 中的二元质量扩散系数,
表示热扩散系数。
对于理想气体,麦克斯韦扩散系数等于二元扩散系数。如果假设外部力对所有组分相同且压力扩散可忽略不计,则 。由于扩散质量通量矢量为 ,上述方程可写为
经过一些数学运算,我们可以得到扩散质量通量矢量 。
其中, 表示组分 的质量分数。其他术语定义如下:
其中,
和 是 矩阵
是广义菲克扩散系数的 矩阵
[649] (第 1094 页)
分子量
用于定义混合物中平均分子量的下标
7.1.1.1.4 扩散系数的动力学理论参数
求解器将使用查普曼-恩斯科格公式 [421] (第 1081 页) 的修正版本来计算
扩散系数 ,采用动力学理论:
地点:
无量纲扩散碰撞积分,这是系统中分子间相互作用的度量。
是作为无量纲温度 的函数来计算的。
其中, 表示温度(以开尔文为单位)。
是玻尔兹曼常数,其定义为气体常数 除以阿伏伽德罗常数。
混合物中的 以开尔文为单位,是几何平均值:
(7.12)
从[248](第1071页)给出的表格值中,可以得出与之间的关系,
而在Ansys Fluent中,这一关系近似为:
对于二元混合物,(以埃为单位,1埃)是根据各个的算术平均值计算的:
7.1.1.1.5 统一路易斯数
所有组分的层流质量扩散率计算如下:
其中, 表示混合物中组分 的质量扩散系数, 是热导率, 是混合物密度, 是混合物的比热容。
7.1.1.1.6 各向异性组分扩散
多孔介质中的各向异性组分扩散模型如下:
- 对于菲克扩散,组分 的质量通量矢量建模为,
其中, 表示孔隙率, 表示多孔区域内的各向异性扩散矩阵,其余符号与公式 7.4(第 239 页)中的定义相同。
- 完整的多元(Maxwell-Stefan)各向异性扩散通量矢量,计算方式类似于公式 7.8(第 240 页),具体如下:
7.1.1.1.7 热扩散系数
热扩散系数可定义为常数、多项式函数、用户自定义函数,或采用以下基于实验数据的与组分相关的表达式,该表达式源自[327](第1076页):
这种形式的Soret扩散系数会导致重分子向加热表面扩散的速度较慢,而轻分子则扩散得更快。
7.1.1.1.8 能量方程中组分传输的处理
对于许多多组分混合流动,由于组分扩散引起的焓传输
会对焓场产生显著影响,不应忽视。特别是,当刘易斯数
对于任何组分,这一比值远非统一,忽视这一项可能导致重大误差。Ansys Fluent 默认会包含这一项。在公式 7.18(第 242 页)中, 表示热导率。
7.1.1.1.9 进口处的扩散
在 Ansys Fluent 的压力基求解器中,进口处组分的净传输包含对流和扩散两个部分。对于密度基求解器,仅包含对流部分。对流部分由用户指定的进口组分质量分数固定。然而,扩散部分取决于进口处计算的组分场的梯度。因此,扩散部分(以及因此的净进口传输)不是事先指定的。有关指定组分净进口传输的信息,请参阅用户指南中的定义单元区域和边界条件以进行组分设置。
7.1.1.2 反应建模的广义有限速率公式
在公式 7.1(第 238 页)中作为源项出现的反应速率,在 Ansys Fluent 中,对于湍流流动,通过以下三种方法之一计算:
-
直接使用有限速率动力学:忽略湍流波动对动力学速率的影响,反应速率直接由广义有限速率化学决定。
-
涡耗散模型:假设反应速率由湍流控制,忽略化学时间尺度的影响,避免了昂贵的阿伦尼乌斯化学动力学计算。该模型计算成本低,但为了得到真实结果,应仅使用一步或两步放热机制。这种方法仅应在整个计算域内关注的化学时间尺度已知相对于湍流时间尺度较快时使用。
-
涡耗散概念(EDC)模型:可以在湍流火焰中结合详细的化学动力学,考虑湍流和动力学的时间尺度。请注意,详细的化学动力学计算可能计算成本较高。
广义有限速率公式适用于广泛的应用场景,包括层流或湍流反应系统,以及具有预混、非预混或部分预混火焰的燃烧系统。
7.1.1.2.1 直接使用有限速率动力学(无TCI)
在没有使用湍流-化学相互作用(TCI)模型的情况下,有限速率动力学通过使用一般反应速率表达式计算化学源项来整合,而不试图明确考虑湍流波动对源项计算的影响。此方法推荐用于层流流动,其中公式是精确的,或者用于使用复杂化学的湍流流动,其中要么预计湍流时间尺度相对于化学时间尺度较快,要么化学足够复杂以至于重要的化学时间尺度高度分散。
由于反应导致的化学组分 的净源项是通过该组分参与的 反应的反应源项之和来计算的。对于多孔介质中的反应,反应导致的净源项是通过将反应速率与孔隙度相乘得到的:
其中,表示组分的分子量,表示孔隙度,而则表示在反应中组分的生成/消耗摩尔速率。反应可能在壁面处的连续相中发生。
考虑以一般形式表示的第个反应,如下所示:
其中,
系统中化学组分的数量
公式7.20(第244页)适用于可逆反应和不可逆反应。对于不可逆反应,逆向反应速率常数为零。
公式7.20(第244页)中的求和针对系统中所有化学组分,但只有作为反应物或产物的组分才具有非零的化学计量系数。
反应中组分的摩尔生成/消耗速率由下式给出:
以下文本的中文翻译:
请注意,在方程7.21(第244页)中,逆反应部分的速率指数是产物组分的化学计量系数。
如需了解CHEMKIN格式中更一般的反应机理信息,请参阅《Ansys Chemkin入门》。关于在Ansys Fluent用户界面中输入化学计量系数和速率指数的信息,包括全局正向(不可逆)反应和基本(可逆)反应,请参阅《Fluent用户指南》中的反应定义输入部分。以下信息涉及在Ansys Fluent中手动输入的机理。
表示第三体对反应速率的净效应。该项由以下公式给出:
其中, 表示第 种物质在第 反应中的第三体效率。默认情况下,Ansys Fluent 在反应速率计算中不包括第三体效应。然而,如果您拥有相关数据,可以选择包含第三体效率的影响。
反应 的正向速率常数 是使用阿伦尼乌斯表达式计算的。
何处,
在Ansys Fluent中定义问题时,您(或数据库)将为提供值,并可选择性地提供。
如果反应是可逆的,默认情况下,反应的反向速率常数将根据正向速率常数通过以下关系计算得出:
其中, 表示第 个反应的平衡常数,其计算方法如下:
其中, 表示大气压(101325 Pa)。指数函数中的项代表吉布斯自由能的变化,其各组成部分的计算方法如下:
在温度和大气压下,和分别表示第种物质的熵和焓。这些值在Ansys Fluent中作为混合物材料的属性进行指定。
Ansys Fluent还提供了在需要时明确指定可逆反应速率参数(前置指数因子、温度指数以及反应的活化能)的选项。在这种情况下,可逆反应的逆向速率常数是根据类似于方程7.23(第245页)的关系式计算的:
何处,
有关指定逆反应速率参数的更多信息,请参阅《Fluent用户指南》中的“反应定义输入”部分。
7.1.1.2.2 压力依赖性反应
通过使用导入的CHEMKIN机制,可以获得压力依赖性反应的一般公式,这些公式的详细描述可以在《Ansys Chemkin入门》中找到。如果您选择通过Ansys Fluent界面手动输入反应表达式,Ansys Fluent可以使用三种方法之一来表示压力依赖性(或压力下降)反应的速率表达式。这些方法是使用导入的CHEMKIN机制可用的公式的一个子集。“下降”反应是指温度和压力使得反应发生在Arrhenius高压极限和低压极限之间,因此不再仅依赖于温度。
在下降区域中,有三种表示速率表达式的方法。最简单的是Lindemann [377](第1079页)形式。还有另外两种相关方法,即Troe方法 [206](第1068页)和SRI方法 [634](第1093页),它们更准确地描述了下降区域。
需要为高压极限和低压极限提供Arrhenius速率参数。然后,将这两个极限的速率系数混合,以产生平滑的压力依赖性速率表达式。在Arrhenius形式中,高压极限 和低压极限 的参数如下:
任意压力下的净速率常数则被视为
其中, 定义为
其中, 表示浴气浓度,可包含第三体效率。若方程 7.31(第 246 页)中的函数 为单位值,则此为 Lindemann 形式。Ansys Fluent 还提供了另外两种描述 的方法,即 Troe 法和 SRI 法。
在Troe方法中,由以下公式给出:
式中
and
参数 和 被指定为输入。
在SRI方法中,混合函数 近似为
式中
除了低压极限 表达式的三个阿伦尼乌斯参数外,您还必须提供 表达式中的参数 和 。
对于化学活化双分子压力依赖反应,任何压力下的净速率常数为
重要提示:化学动力学机制通常包含广泛的时间尺度,并形成一组高度非线性、刚性的耦合方程。有关求解过程的指南,请参阅用户手册中的化学混合和有限速率化学的求解过程。此外,如果您有CHEMKIN [300](第1074页)格式的化学机制,可以将其导入Ansys Fluent。请参阅用户手册中的导入体积动力学机制的CHEMKIN格式部分。
7.1.1.2.3 涡耗散模型
在某些燃烧条件下,燃料燃烧迅速,整体反应速率受湍流混合控制。例如,在高温非预混火焰中,湍流缓慢地将燃料和氧化剂对流传输/混合到反应区,在那里它们迅速燃烧。在某些预混火焰中,湍流缓慢地将冷反应物和热产物对流传输/混合到反应区,反应在那里迅速发生。在这种情况下,一种近似方法是假设燃烧受混合限制,从而忽略复杂的化学动力学速率,转而假设混合后立即燃烧。
对于“混合即燃烧”的近似,Ansys Fluent 提供了一种基于 Magnussen 和 Hjertager [410](第1080页)工作的湍流-化学相互作用模型,称为涡耗散模型。使用此模型,由于反应 产生的组分 的净生成速率 ,由以下两个表达式中较小的一个(即限制值)给出:
其中,
任意产物组分的质量分数,
特定反应物的质量分数,
经验常数,等于4.0
经验常数,等于0.5
在公式7.39(第247页)和公式7.40(第247页)中,化学反应速率受大涡混合时间尺度 / 控制,如Spalding [624](第1093页)的涡破碎模型所述。只要存在湍流 ,燃烧就会进行,不需要点火源来启动燃烧。这对非预混火焰通常是可以接受的,但在预混火焰中,反应物一进入计算域就会燃烧,位于火焰稳定器上游。为了解决这个问题,Ansys Fluent提供了有限速率/涡耗散模型,其中同时计算有限速率反应速率(公式7.21(第244页))和涡耗散速率(公式7.39(第247页)和公式7.40(第247页))。净反应速率取这两个速率中的最小值。实际上,有限速率动力学起到了动力学“开关”的作用,防止在火焰稳定器之前发生反应。一旦火焰被点燃,涡耗散速率通常小于阿伦尼乌斯速率,反应受混合限制。
重要提示:
-
尽管Ansys Fluent允许在涡耗散和有限速率/涡耗散模型中使用多步反应机理(反应数目大于2),但这些模型很可能会产生不正确的解。原因在于,多步化学机理通常基于Arrhenius速率,每个反应的速率各不相同。在涡耗散模型中,每个反应都具有相同的湍流速率,因此该模型仅适用于单步(反应物→产物)或两步(反应物→中间体,中间体→产物)的总体反应。该模型无法预测如自由基等动力学控制的组分。要在湍流流动中结合多步化学动力学机理,请使用EDC模型(详见《LES的涡耗散模型》(第248页))。
-
涡耗散模型要求产物启动反应(参见公式7.40(第247页))。在为稳态流动初始化解时,Ansys Fluent将所有组分的质量分数设置为用户指定的初始值中的最大值和0.01。这通常足以启动反应。然而,如果你首先收敛一个混合解,其中所有产物的质量分数为零,那么你可能需要在反应区中修补产物以点燃火焰。详细信息请参阅用户指南中的“稳态燃烧模拟中的点火”。
7.1.1.2.4 LES的涡耗散模型
当使用LES湍流模型时,公式7.39(第247页)和公式7.40(第247页)中的湍流混合速率 / 被替换为亚格子尺度混合速率。该速率计算如下:
哪里,
7.1.1.2.5 涡耗散概念(EDC)模型
涡耗散概念(EDC)模型是涡耗散模型的一种扩展,它允许在湍流流动中包含详细的化学机理[409](第1080页)。该模型假设反应发生在称为精细尺度的小湍流结构内。这些精细尺度占据计算单元的一小部分,其特征是体积分数 。在精细尺度上的燃烧被假设为恒压反应器,初始条件取为单元内的当前组分和温度。反应过程在时间尺度 上进行,受方程7.21(第244页)的反应速率控制,并使用常微分方程(ODE)求解器进行数值积分。守恒方程中组分 的源项,如方程7.1(第238页)所示,被建模为
其中, 是在时间尺度 上经过反应后的细尺度组分质量分数。
EDC 模型可以将详细的化学机理纳入湍流反应流动中。然而,典型的机理通常是刚性的,其数值积分在计算上成本较高。因此,只有在快速化学假设无效的情况下才应使用该模型,例如模拟低温或高压燃烧、快速淬灭火焰中的缓慢 CO 燃烧,或选择性非催化还原(SNCR)中的 NO 转化。
有关使用 EDC 模型获取解决方案的指南,请参阅用户指南中的“刚性化学系统的解决方案”部分。
7.1.1.2.5.1 标准 EDC 模型
在标准 EDC 模型 [217](第 1069 页)中,反应性细尺度的体积分数被建模为:
其中长度尺度 的计算如下:
哪里
运动粘度
湍流动能
湍流动能耗散率
积分时间尺度 被建模为:
其中, 是一个时间尺度常数,等于 0.4082。
7.1.1.2.5.2 部分搅拌反应器 EDC 模型
标准 EDC 模型(详见标准 EDC 模型 (第 249 页))已成功应用于通过结合详细的化学机理来模拟湍流-化学耦合。然而,标准 EDC 模型的主要缺点是长度尺度常数 和时间尺度常数 都不依赖于局部流动和化学变量。研究表明,标准 EDC 模型存在局限性。例如,当应用于模拟中等和强烈低氧稀释(MILD)系统(由于强氧稀释导致的慢化学反应)时,EDC 模型会显著高估温度水平 [361](第 1078 页),[362](第 1078 页)。
一些作者提出了 EDC 模型的广义化,以应用于更广泛的火焰区域。部分搅拌反应器(PaSR)模型 [176](第 1067 页),[720](第 1098 页)已被应用于温和和超音速燃烧。它在概念上类似于标准 EDC 模型,但具有不同的反应体积分数和时间尺度的定义:
在公式7.46(第250页)中,的定义如下:
在Ansys Fluent中,化学时间尺度 是根据[166](第1066页)中概述的方法计算的:
其中, 表示 、、、 和 的反应速率(单位为 ),这些较慢的主要组分主导了精细时间尺度和更大的火焰结构。
混合时间尺度 被视为湍流积分时间尺度 的一部分:
其中,可以是常数值,也可以是局部湍流雷诺数的函数。
其中, 是 湍流模型中的常数。
当使用方程7.51(第250页)来计算 时,该模型被称为分形方法 [176](第1067页)。指数系数 表示为:
其中, 表示分形维数。当 时,对应于 Kolmogorov 时间尺度;而当 时,则对应于积分时间。
需要注意的是,当使用 LES 湍流模型时, 是通过 Kolmogorov 时间尺度和湍流时间尺度的几何平均值计算得出的 [366](第 1078 页)。
7.1.1.2.6 加厚火焰模型
预混火焰的典型层流火焰厚度约为毫米级。由于层流火焰传播速度由火焰内的组分扩散、热传导和化学反应决定,因此需要足够的网格分辨率来预测正确的层流火焰速度。相比之下,燃烧室尺寸通常远大于层流火焰厚度,即使使用非结构化和解自适应网格,也无法经济地解析火焰。
预混层流火焰速度,记为 ,与 成正比,其中 是扩散系数, 是反应速率。层流火焰厚度与 成正比。因此,通过按比例增加扩散系数并减少反应速率,可以人为地增加层流火焰厚度,而不改变层流火焰速度。这样,加厚的火焰就可以在粗网格上得到可行解析,同时仍能捕捉到正确的层流火焰速度。
在 Ansys Fluent 中,加厚因子 的计算方法如下:
其中, 表示网格尺寸, 是层流火焰厚度,而 则是用户指定的火焰中的点数(默认为8)。网格尺寸 由 确定,其中 是单元体积, 是空间维度(2或3)。层流火焰厚度 由用户输入,可以是常数、用户定义函数,或者计算为 ,这里的 是热扩散系数,计算方式为 (其中 是热导率, 是密度, 是比热容)。
所有组分的扩散系数以及热导率都乘以加厚因子 ,而所有反应速率则除以 。然而,在火焰之外,这些增强的扩散性可能导致错误的混合和热传递,因此火焰仅在反应前沿周围的狭窄带内动态加厚。这一带宽是通过将 乘以一个因子 来计算的, 的计算方法为...
在公式7.54(第251页)中, 是反应速率的空间滤波绝对值,而 是一个默认值为10的常数。绝对反应速率经过多次滤波, 是域中 的最大值。 的范围从火焰带附近的单位值到该带外部的零值。
厚火焰模型(TFM)[497](第1086页)通常与单步化学机理一起使用,其中全局反应已调整以提供正确的层流火焰速度。原则上,TFM可以与多步反应机理一起使用,但所有成分分布应在火焰内得到充分解析。TFM在基于压力和基于密度的求解器中均可用。
虽然TFM可用于模拟层流火焰,但其最常见的应用是作为湍流预混和部分预混火焰的LES燃烧模型。预混火焰的湍流火焰速度主要由火焰褶皱决定,这增加了火焰表面积。
火焰的增厚改变了其与湍流的相互作用。这应在计算组分形成和破坏速率时考虑。随着火焰变厚,湍流褶皱火焰的能力减弱,这影响了湍流火焰速度的传播。湍流化学相互作用的减少通过将反应速率乘以效率函数 来补偿。在Ansys Fluent中,Charlette和Colin效率函数可用于模拟这种现象。
Colin效率函数通过以下公式计算:
其中,
= 使用Durand和Polifke[154](第1065页)提出的方法计算的次网格尺度湍流速度。
= 对应于加厚火焰的滤波宽度,通常比物理网格长度尺度大一个数量级。默认值取为局部单元网格间距的10倍。
= 层流火焰速度。
= 由公式7.56(第252页)计算的参数。
和 = 由公式7.58(第253页)定义。
其中,
用户定义的参数,其数量级为1。
修正参数,取值范围从0到1。该修正基于局部湍流条件和截止长度值,截止长度是指低于此长度尺度时,涡流不会改变火焰的最小长度尺度。截止长度根据厚化火焰厚度和雷诺数计算得出。
是一个常数,值为0.28。
方程7.56(第252页)中的湍流雷诺数 基于积分长度尺度 和积分长度尺度上的均方根速度波动。积分长度尺度是流场中最大涡流的长度,它依赖于几何形状,通常使用特征尺寸(如燃烧器直径、入口直径或钝体尺寸)的 到 作为其值。积分长度尺度上的均方根速度波动计算如下:
其中, 表示在积分长度尺度 上的均方根速度。
在这里,上标0表示未经加厚的火焰,而上标1表示加厚的火焰。
当流动为层流时,等于0,此时Colin效率函数简化为1。当湍流速度波动变化时,即...
科林效率函数趋近于一个理论上的最大极限:
查尔莱特效率函数[100](第1062页),[687](第1097页)的计算方法如下:
对应于加厚火焰的过滤器宽度,通常比物理网格长度尺度大一个数量级。默认值是局部单元网格间距的10倍。
层流火焰厚度。
次网格尺度湍流速度。
层流火焰速度。
过滤器尺度下的次网格雷诺数。
指数设定为常数值0.5。
的计算公式为:
当 ( b = 1.4 ) 时,
在上述方程中, 是值为1.5的科尔莫戈罗夫常数。
所有组分的反应速率都乘以 。每个单元内的有效组分扩散系数(以及热导率)是动态确定的,具体如下:
其中, 表示分子(层流)扩散系数,而 表示湍流扩散系数。 可以通过 Ansys Fluent 中提供的任何方法计算,包括动力学理论和用户定义函数(UDFs)。由于一步反应的动力学系数总是调整以捕捉实际的层流火焰速度,因此在进行 TFM 模拟时应使用在此调整模拟中使用的相同传输属性。在火焰附近的狭窄带中,当 为一时,湍流扩散系数被关闭,分子扩散系数增强了一个因子 EF。远离火焰的地方,当 为零时,有效扩散系数是未增厚的 值。
7.1.1.2.7 松弛至化学平衡模型
当使用有限速率(FR)化学模型,或如上所述使用涡耗散(ED)、有限速率/涡耗散(FR/ED)或 EDC 模型时,化学组分根据规定的动力学机制演变。在松弛至化学平衡模型中,组分的组成被驱动至其平衡状态。第 平均组分守恒方程(方程 7.1(第 238 页))中的反应源项被建模为
何处,
组分 的平均质量分数
上标 表示化学平衡状态
特征时间尺度
公式 7.68(第 254 页)表明,组分在特征时间 内向其化学平衡状态反应,如特征时间模型 [320](第 1075 页)所述。由于化学平衡不依赖于反应或反应速率,对于给定的 ,公式 7.68(第 254 页)中的反应源项与反应机理无关。
化学平衡松弛模型是适用于任何湍流-化学相互作用选项的一种选择。当不使用湍流-化学模型时,特征时间按以下方式计算:
其中, 表示细胞内的对流/扩散时间尺度,而 则代表细胞化学反应的时间尺度,该尺度是通过模型来描述的。
在公式7.70(第255页)中,是一个模型常数(默认值为1),下标表示用户指定的燃料组分,是燃料组分的质量分数,是燃料组分质量分数的变化率,而min表示所有指定燃料组分中的最小值。因此,代表燃料组分的化学点火时间尺度,用于防止预混火焰自燃,类似于FR/ED模型。需要注意的是,评估需要一个动力学机制。
对于ED模型,特征时间尺度评估为,其中湍流时间尺度为,默认的湍流速率常数为。对于FR/ED模型,特征时间尺度计算为。通常,ED和FR/ED模型采用一步反应,或者两步反应,其中烃类热解为CO,然后氧化为。化学平衡松弛模型可以视为ED类型模型的扩展,其中组分向化学平衡反应而不是完全反应。该模型应能更准确地预测中间组分,如和建模所需的自由基,如和。
由于化学平衡计算通常比详细化学模拟的计算成本低,化学平衡松弛模型可用于为全动力学稳态模拟提供良好的初始条件。该模型也可替代涡流耗散模型,其中解趋向化学平衡而不是完全反应。
化学平衡假设在烃类燃料的富区可能导致较大误差。Ansys Fluent 提供了通过增加富区中特征时间尺度 来降低方程 7.68(第254页)中反应速率的选项。使用此选项,单元格内的局部当量比会被计算为
其中,和分别表示氧、氢和碳的原子摩尔分数。当局部当量比超过指定的富当量比时,时间尺度会乘以一个因子。默认的富当量比为1,默认的指数因子为2。文本界面中提供了在富混合物中启用慢反应速率的选项。
由于化学平衡计算可能会消耗计算资源,因此推荐(且默认)使用ISAT表格法。随着表格中条目数量的增加,初始的Ansys Fluent迭代速度会显著加快,大多数对表格的查询都是通过插值完成的。由于化学平衡组成仅由初始温度、压力和混合物的元素组成决定,因此ISAT表格的维度对于等压系统为。在大多数燃烧情况下,元素数量远小于化学组分数量,因此ISAT表格构建相对较快。也可以选择直接积分选项,此时将在所有单元格中始终执行平衡计算。
7.1.1.3 使用两温度模型的有限速率化学
在超音速情况下,如解离和重组等化学反应可能对流动产生重大影响。如果流动特征时间和化学反应时间大致相同,流体单元在某一位置停留的时间不足以使局部化学反应达到平衡,因此流动可能处于局部化学非平衡状态。两温度模型可以与有限速率化学模型结合使用,以模拟这些热化学非平衡流动。
当双温度模型与有限速率化学模型耦合,用以描述高超音速流动中的热化学非平衡现象时,方程7.23(第245页)的形式变为:
其中, 是控制温度。对于解离反应,系数 ,以考虑解离反应受到振动模式极大影响的事实。对于交换反应,系数 。
有关指定控制温度系数的详细信息,请参阅 Fluent 用户指南中的“使用双温度模型”部分。
7.1.2 壁面表面反应与化学气相沉积
对于气相反应,反应速率是以体积为基础定义的,化学组分的产生和消耗净速率成为组分守恒方程中的源项。对于表面反应,吸附和解吸的速率既受化学动力学控制,也受向表面和从表面的扩散控制。因此,壁面表面反应在气相中以及在反应表面上都会产生化学组分的源和汇。
本节介绍了关于壁面表面反应和化学气相沉积的理论信息。相关信息可以在以下章节中找到:
- 7.1.2.1. 表面覆盖率对反应速率的修正
- 7.1.2.2. 表面化学的反应-扩散平衡
- 7.1.2.3. 低压气体系统的滑移边界条件公式
关于如何使用壁面表面反应和化学气相沉积的更多信息,请参阅用户指南中的“壁面表面反应与化学气相沉积”部分。
考虑以一般形式书写的第 壁面表面反应如下:
其中,、 和 分别代表气相组分、体相(或固态)组分以及表面吸附(或位点)组分。、 和 是这些组分的总数。、 和 表示每个反应物组分 的化学计量系数,而 、 和 则是每个生成物组分 的化学计量系数。 和 分别是反应的前向和后向速率。
公式 7.73(第 256 页)中的求和涵盖了系统中所有的化学组分,但只有作为反应物或生成物的组分才具有非零的化学计量系数。因此,未参与反应的组分将从方程中消失。
第 个反应的速率为...
其中,[ ] 表示壁面上表面吸附组分的摩尔浓度。 和 分别是反应中作为反应物和产物的第 种气态组分的速率指数。变量 和 分别是反应中作为反应物和产物的第 种位点组分的速率指数。假设反应速率不依赖于体相(固体)组分的浓度。由此,每个组分 的净摩尔生成或消耗速率由以下公式给出:
反应的正向速率常数是利用阿伦尼乌斯方程计算得出的。
何处,
您(或数据库)将为以下变量提供数值:, 以及 。
若要考虑质量传递效应并模拟热释放,请参阅用户指南中的“连续性方程中包括表面质量传递”、“能量方程中的壁面表面质量传递效应”以及“模拟壁面表面反应引起的热释放”部分。
如果反应是可逆的,反应 的逆向速率常数 将根据正向速率常数通过以下关系计算得出:
其中, 表示第 个反应的平衡常数,由...计算得出。
其中, 表示大气压(101325 Pa)。指数函数内的项代表吉布斯自由能的变化,其组成部分根据公式7.26(第245页)和公式7.27(第245页)计算。
表示不同类型站点的数量, 是站点类型 的站点密度。 和 分别是反应 中类型 的第 站点组分的化学计量系数。
7.1.2.1 表面覆盖反应速率修正
Ansys Fluent 提供了根据组分站点覆盖度来修正表面反应速率的选项。在这种情况下,第 反应的正向速率常数评估如下:
在方程7.81(第258页)中,反应中组分的三个表面覆盖率修正参数为、和。对于非表面速率修正的反应组分,这些参数默认值为零。表面(覆盖)位分数是指由组分覆盖的表面位点的比例,其定义为:
其中, 表示表面位点浓度, 为表面位点密度(参见方程 7.87,第259页)。
7.1.2.2 表面化学的反应-扩散平衡
表面反应会改变气相、表面吸附(位点)以及体相(固体)组分。在反应表面上,由于扩散和流动引起的每种气体组分的流向表面的质量通量与表面上的消耗/生成速率达到平衡。
壁面处的质量分数 与浓度有关,
表示由于表面反应引起的质量沉积或刻蚀的净速率;也就是说,
表示壁面处的组分浓度,其定义为
其中, 表示站点密度,而 则代表组分 的站点覆盖度。
方程 7.83(第 258 页)和方程 7.84(第 258 页)通过逐点耦合的牛顿求解器来求解依赖变量 和 。当牛顿求解器无法收敛时,方程 7.83(第 258 页)和方程 7.84(第 258 页)将在 ODE 求解器中通过时间推进法求解,直至达到收敛。若 ODE 求解器也失败,则禁用反应-扩散平衡,假设 等于单元中心值 ,并在 ODE 求解器中仅推进站点覆盖度 直至收敛。你可以通过文本界面命令 define/models/species/reaction-diffusion-balance 手动禁用反应-扩散平衡。
随后,有效的气相反应源项便可用于求解气相组分传输方程 7.1(第 238 页)。
方程7.83(第258页)中的扩散项计算为单元中心与壁面中心处的组分质量分数之差,再除以这两个中心点之间的法向距离。Ansys Fluent通过引入表面面积涂层因子来模拟发生在催化剂结构内部的反应。通常,涂层是一种载体,催化材料悬浮其中。与裸露基底相比,这些材料通过形成粗糙和不规则的表面,增加了表面积;从而增加了可供表面反应的催化活性反应面积。为了利用可用的反应表面,Ansys Fluent提供了定义涂层因子的选项。该系数乘以反应表面,计算出可用的反应表面,从而线性修正表面反应速率。Ansys Fluent还可以使用多孔介质模型对未解析壁面上的表面化学进行建模。该模型适用于催化管束或多孔泡沫矩阵,在这些情况下解析单个壁面是不可行的。当在多孔单元区域启用表面反应时,必须指定表面与体积比。方程7.83(第258页)中扩散项所需的壁面法向距离计算为表面与体积比的倒数。
7.1.2.3 低压气体系统的滑移边界条件
大多数半导体制造设备在远低于大气压的条件下运行,通常只有几毫托。在这种低压下,流体流动处于滑移状态,通常使用的无滑移速度和温度边界条件不再有效。
克努森数,记作 ,定义为平均自由程与系统特征长度尺度的比值,用于量化连续流态。由于平均自由程随压力降低而增加, 值的高端代表自由分子流,低端则代表连续流态。介于这两个极端之间的范围被称为滑移区(0.01 < < 0.1)[63](第1060页)。在滑移区,气体在固体表面的速度与壁面运动速度不同,且表面处的气体温度与壁温也不同。为了简洁有效,Ansys Fluent 采用了麦克斯韦模型来描述这些物理现象。
- 速度滑移
在此, 与 分别代表沿壁面切向和法向的速度分量。下标 、 和 分别指代气体、壁面以及单元中心的速度。 表示从单元中心到壁面的距离。 是特征长度。 为气体混合物的动量适应系数,其值根据系统中各气体组分的质量分数加权平均计算得出。
平均自由程 的计算方法如下:
是组分 的 Lennard-Jones 特征长度。 是玻尔兹曼常数,值为 。
方程 7.88(第 260 页)和方程 7.89(第 260 页)表明,尽管气体垂直于壁面的速度分量与壁面法向速度相同,但其切向分量会发生滑移。这些值介于单元中心值和壁面值之间。这两个方程可以结合,得到一个广义的表达式:
何处
温度突变
或者等价地
哪里
是气体混合物的热适应系数,其计算公式为 。请注意:
低压滑移边界条件公式仅在使用基于压力的求解器时可用。
7.1.3 颗粒反应
以下各节提供了有关颗粒反应的理论信息:
- 7.1.3.1. 燃烧颗粒表面反应
- 7.1.3.2. 含化学反应的多组分颗粒
7.1.3.1. 燃烧颗粒表面反应
如多重表面反应模型(第484页)所述,可以定义多个颗粒表面反应来模拟燃烧离散相颗粒的表面燃烧过程。本节提供了关于颗粒表面反应的理论背景。相关信息可在以下各节中找到:
- 7.1.3.1.1. 一般描述
- 7.1.3.1.2. Ansys Fluent模型公式
- 7.1.3.1.3. 多气体相反应物的化学计量学扩展
- 7.1.3.1.4. 固-固反应
- 7.1.3.1.5. 固体分解反应
- 7.1.3.1.6. 固体沉积反应
- 7.1.3.1.7. 颗粒表面上的气态固体催化反应
更多关于使用颗粒表面反应的信息,请参阅用户指南中的燃烧颗粒表面反应。
7.1.3.1.1 一般描述
计算焦炭颗粒燃烧速率的关系式由Smith [609](第1092页)详细提出并讨论。颗粒反应速率 可以表示为
其中,
体积扩散系数
体积中反应气体组分的平均浓度
颗粒表面反应气体组分的平均浓度
化学反应速率系数(单位不定)
表观反应级数(无量纲)
在公式 7.98(第 261 页)中,颗粒表面的浓度 是未知的,因此需要将其消去,并将表达式重写如下:
这个方程需要通过迭代过程来求解,除了或的情况。当时,方程7.99(第262页)可以写成
在的情况下,如果颗粒表面存在有限的反应物浓度,固体消耗速率等于化学反应速率。如果表面没有反应物,固体消耗速率会突然转变为扩散控制速率。然而,在这种情况下,出于稳定性考虑,Ansys Fluent将始终使用化学反应速率。
7.1.3.1.2 Ansys Fluent模型公式
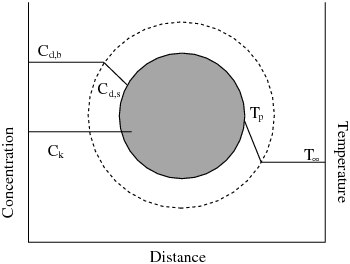
图7.1(第262页)以示意图形式展示了一个在气相中经历放热反应的颗粒,即多表面反应模型中的反应颗粒。和是公式12.159(第485页)中的温度。
图7.1:多表面反应模型中的反应颗粒
基于上述分析,Ansys Fluent使用以下方程来描述颗粒表面组分与气相组分的反应速率。在这种情况下,反应的化学计量关系由...
反应速率表示为
其中,
表示颗粒表面组分消耗速率
表示颗粒表面积
表示颗粒中表面组分 的质量分数
表示有效因子(无量纲)
表示单位面积上颗粒表面组分反应速率
表示气相组分的体积分压 (Pa)
表示反应 的扩散速率系数
表示反应 的动力学速率(单位不定)
表示反应 的表观反应级数
有效因子 与表面积相关,可用于多反应情况下的每个反应。 由以下公式给出:
反应速率 定义为
对于反应级数 ,颗粒表面组分消耗速率的表达式为:
对于反应级数 的情况:
7.1.3.1.3 多气体反应物的化学计量扩展
当反应中涉及多个气体相反应物时,反应的化学计量必须进行扩展以适应这种情况:
粒子组分 + 气体相组分1 + 气体相组分2 + ...
为了描述在存在种气相组分的情况下,粒子表面组分的反应速率,必须为每个固体粒子反应定义扩散限制组分,即在体相和粒子表面之间浓度梯度最大的组分。对于其余组分,假定表面和体相的浓度相等。扩散限制组分的浓度在图7.1:多重表面反应模型中的反应粒子(第262页)中显示为和,而所有其他组分的浓度则表示为。对于具有多种气相反应物的化学计量,方程7.103(第262页)和方程7.106(第263页)中的体相分压是反应的扩散限制组分的体相分压,记为。
随后,反应的动力学速率定义为
何处,
启用此模型后,常数 (方程 7.104,第 263 页)和有效因子 (方程 7.102,第 262 页)需在反应对话框中输入(参见用户指南中的颗粒表面反应用户输入部分)。
7.1.3.1.4 固-固反应
只要颗粒表面反应物和产物存在于同一颗粒上,就可以对仅涉及颗粒表面反应物的反应进行建模。
此情况下的反应速率由公式7.107(第263页)给出。
7.1.3.1.5 固体分解反应
颗粒表面组分的分解反应可以被建模。
对于这种情况,反应速率由方程7.102(第262页)至方程7.109(第264页)给出,其中扩散受限的组分现在是反应的气态产物。如果反应中存在多个气态产物组分,则需要将颗粒反应的扩散受限组分定义为块体与颗粒表面之间浓度梯度最大的组分。
7.1.3.1.6 固体沉积反应
在颗粒上沉积固体组分的反应可以基于以下假设进行建模:
理论分析和公式7.102(第262页)至公式7.109(第264页)应用于表面反应速率计算,其中在公式7.102(第262页)、公式7.106(第263页)和公式7.107(第263页)中,表面组分的质量分数设为统一。
在Ansys Fluent中,为了使颗粒表面的组分沉积在颗粒上,颗粒中必须已经存在该组分的一定质量。这使得沉积反应可以选择性地激活特定的注入颗粒。因此,为了在颗粒上启动固态组分沉积反应,必须在设置注入属性对话框(或设置多个注入属性对话框)中定义颗粒包含待沉积固态组分的微量质量分数。有关定义颗粒表面组分质量分数的详细信息,请参阅用户指南中的“使用多表面反应模型进行离散相颗粒燃烧”部分。
7.1.3.1.7 颗粒表面上的气固催化反应
颗粒表面上的气态组分催化反应也可以按照公式7.102(第262页)至公式7.109(第264页)进行表面反应速率计算,其中在公式7.102(第262页)、公式7.106(第263页)和公式7.107(第263页)中,表面组分的质量分数设为统一。要应用此类反应,请参阅用户指南中的“气固催化反应建模”部分。有关定义颗粒表面组分质量分数的详细信息,请参阅用户指南中的“使用多表面反应模型进行离散相颗粒燃烧”部分。
7.1.3.2 含化学反应的多组分颗粒
多组分颗粒的组分可以参与化学反应。反应可以在颗粒组分之间发生,或者与气相组分发生,颗粒组分允许依次或同时蒸发和反应。有几种反应模型作为内置功能可用,同时还有用户定义选项。多组分颗粒可能包含参与反应的、挥发性的或惰性的组分,这些惰性组分不参与任何反应。
与燃烧颗粒的固态组分类似,多组分颗粒的组分可以参与如下所述的化学反应:
-
颗粒组分与一个或多个气相组分反应
-
颗粒组分与一个或多个颗粒组分反应
-
颗粒组分分解为气相组分
-
气相组分转化为颗粒组分并沉积在颗粒上
-
催化反应,其中气相组分在颗粒表面反应而不消耗颗粒组分
对于这些反应类型,Ansys Fluent 使用以下反应模型:
- 动力学/扩散
反应速率根据 Ansys Fluent 模型公式(第262页)计算。
- 表面动力学
反应速率计算如下:
其中:
颗粒组分 的反应 的速率
颗粒的表面积 。
有效因子。
反应速率系数 。
颗粒组分 的质量分数。
气体反应物 的分压。
气体反应物 的反应级数。
反应速率系数 表示为:
其中:
指前因子。该因子必须以与用于基于 的反应速率的单位一致的国际单位制(SI)单位来指定。
颗粒的温度(K)。
温度指数。
活化能(J/kg-mol/K)。
理想气体常数。
体积动力学反应速率计算如下:
其中:
颗粒组分 的反应 的速率
颗粒体积
颗粒组分 的分子量
反应速率系数。反应速率系数的表达形式与方程 7.113(第 266 页)相同,其中指前因子 的单位以 kg-mol 为基准。
和 气体反应物 和颗粒反应物 的浓度,分别为
和 气体反应物 和颗粒反应物 的反应级数。
- 相变
在此反应模型中,颗粒组分可以与其他反应组分同时蒸发。反应计量中只允许一种固态/凝聚态颗粒物质作为反应物,以及一种气体物质作为产物。颗粒组分 向气体组分 的相变速率计算如下:
颗粒组分的相变速率
颗粒表面积
颗粒组分的分子量
质量传递系数
和 颗粒反应物在颗粒表面和气相组分的浓度,分别为
- 热解(仅限自由流颗粒)
其中:
粒子组分 的反应 的反应速率
指前因子(以 为基准的 单位)。
粒子直径 。
粒子温度(K)。
温度指数。
活化能(J/kg-mol/K)。
理想气体常数。
除了上述反应模型外,您还可以使用用户定义函数来定义自己的反应速率。有关使用这些模型的更多信息,请参阅 Fluent 用户指南中的“含化学反应的多组分粒子”部分。
7.1.4 电化学反应
本节介绍了在 Ansys Fluent 中建模电化学反应的理论背景。以下主题将进行讨论:
7.1.4.1. 概述
7.1.4.2. 电化学反应模型理论
7.1.4.1 概述
电化学电荷转移反应是一种涉及中性组分、带电离子和电子的化学反应。与电化学相关现象包括电池、燃料电池、腐蚀和电沉积。电荷转移反应发生在电极(阳极或阴极)与电解质之间的相界面,这一界面被称为法拉第表面。
7.1.4.2 电化学反应模型理论
第 个电化学反应的一般形式为:
其中:
表示液体或固体组分
是组分总数
和 分别是反应 中作为反应物和产物的第 组分的化学计量系数
是第 组分的电荷数
表示电子
对于中性组分,电荷数 为零。
在 Ansys Fluent 中,法拉第界面处的电化学反应 的电荷转移反应速率由 Butler-Volmer 方程确定:
或通过其使用Tafel斜率的替代形式:
其中:
法拉第电流密度
交换电流密度
化学组分总数
活化能
通用气体常数 (J/kmolK)
温度 (K)
参考温度 (K)
反应 中组分 的摩尔浓度
反应 中组分 的参考摩尔浓度
组分 的速率指数
阳极电荷转移系数
阴极电荷转移系数
阳极反应的塔菲尔斜率 (V)
阴极反应的塔菲尔斜率 (V)
法拉第常数 (C/kmol)
过电位 (V)
注意:
请注意,在公式 7.118 (第 268 页) 中, 和 是电荷转移系数,而不是文献中常用于巴特勒-沃尔默方程的对称因子。
过电位 的计算公式为:
其中, 和 分别表示电极和电解质电位(V),而 表示平衡电位(V)。
如公式 7.117(第 267 页)所示的一般反应,在正向和反向同时发生。阳极反应发生在产生电子的方向,而阴极反应发生在消耗电子的方向。
公式 7.118(第 268 页)中的第一项表示阳极方向的速率,而第二项表示阴极方向的速率。这两者之间的差异给出了反应的净速率。反应的净方向取决于表面过电位的符号。如果过电位为正,整体反应向阳极反应方向进行;如果为负,则向阴极反应方向进行。
一般反应(公式 7.117(第 267 页))还表明,在电化学反应中,电流与组分的生成/消耗速率成正比。对于第 反应中的组分 ,这种相关性由法拉第定律表达:
其中, 表示组分的生成/消耗速率, 代表组分的分子量,而 则是电化学反应 产生的总电子数,计算方法如下:
由于所有电化学反应,组分 的总生成/消耗速率可计算如下:
根据法拉第定律(见方程7.121,第269页),电化学反应的速率可以用电流或物质质量变化来表示,因为这两个量是直接成比例的。
根据巴特勒-沃尔默方程(见方程7.118,第268页),电化学反应的驱动力是电极-电解质界面上的电位差。介质中的电势可以从电荷守恒定律推导出来:
其中, 表示电流密度矢量。
在固相中,电流密度与电势梯度之间的关系遵循欧姆定律:
其中, 表示电导率, 表示电势。
因此,电势场受拉普拉斯方程支配:
在液相(或电解质相)中,电流是带电粒子的净通量:
其中:
由组分扩散、组分对流以及在电场中组分(固态或液态)迁移产生的组分 的通量密度
流场的速度
组分 的浓度
组分 的扩散系数
组分 的迁移率
一般来说,电化学理论的处理过于复杂,难以实际应用。为了简化公式 7.127(第 270 页),通常假设电荷中性:
这消除了公式7.127(第270页)中的第二项。
此外,公式7.127(第270页)中的第一项通常被认为与最后一项相比可以忽略不计。对于充分混合或电解质浓度高的混合物来说,这是一个合理的假设。因此,公式7.127(第270页)中仅剩下最后一项。
定义离子电导率为,
电荷守恒方程(方程7.127,第270页)简化为:
因此,在固相区和液相区均求解相同的拉普拉斯方程。
电荷中性并未被明确强制执行。相反,假设存在未表示的等量反向电荷的组分,这些组分会抵消已表示组分的电荷。
电场可能对电解质中的带电组分施加力,这导致组分传输方程中增加了一个额外项(见方程7.1,第238页)。
电化学能为能量方程(方程5.1,第172页)贡献两种独特的热源。
第一种热源源于法拉第界面处的电荷转移反应,其模型表示为:
其中, 表示法拉第电流密度,而 则是过电位(参见公式 7.120,第 269 页)。
第二个项是由于电荷运动引起的焦耳热:
7.1.5. 反应通道模型
本节提供关于反应通道模型的理论背景。相关信息可在以下章节中找到:
- 7.1.5.1. 概述
- 7.1.5.2. 反应通道模型理论
如需了解更多关于使用反应通道模型的信息,请参阅用户指南中的反应通道模型部分。
7.1.5.1 概述
某些流动几何包含长而细的通道(也称为管道或管道),其中含有与外部流体进行热交换的反应流体。这类壳管配置的例子包括燃料重整器和裂化炉。通道内的流动迅速建立稳定的、完全发展的径向分布。在这些通道中,化学组分和温度的演变可以通过低维模型进行准确建模,通常假设为抛物线流动。这比在通道内部解析网格并使用三维求解器同时处理通道内外流动和化学反应要高效得多。Ansys Fluent中的反应通道模型允许对具有简单流动但发生在长而细通道中的复杂化学反应进行经济实惠的模拟,这些通道与几何结构复杂的外部流动(具有简单或无化学反应)热耦合。
关于反应通道模型的限制,请参阅Fluent用户指南中的反应通道模型概述和限制部分。
7.1.5.2 反应通道模型理论
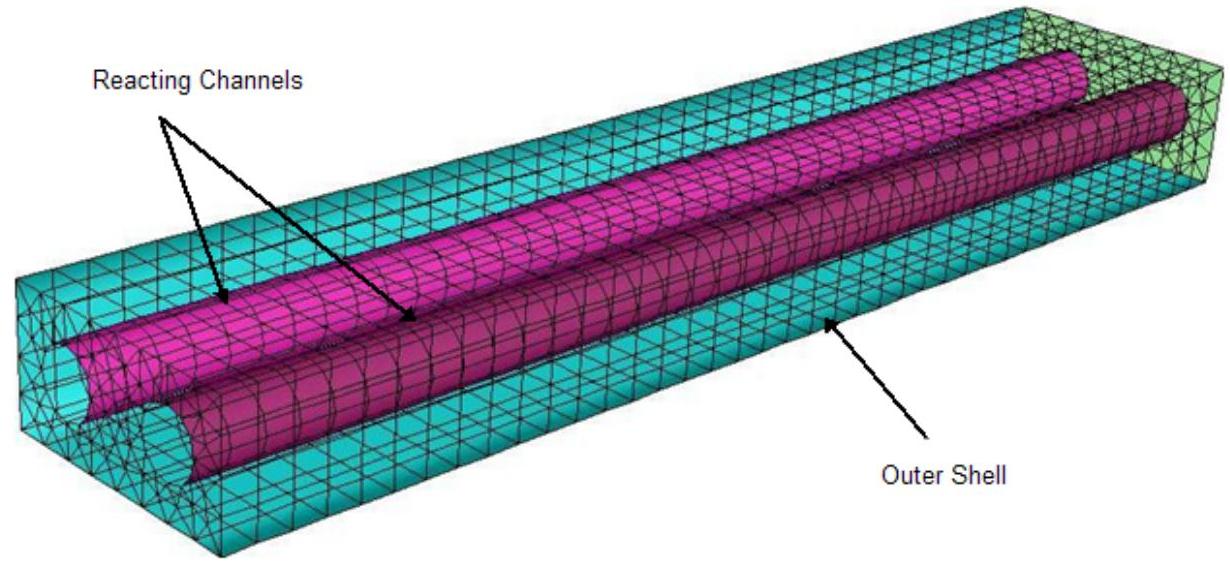
在反应通道模型中,所有通道壁必须在三维空间中进行几何解析。然而,只有外壳的几何形状需要进行网格划分;通道壁内部不应包含任何单元。图7.2(第272页)展示了一个典型配置,包含两个圆形通道及其周围的外壳。通道内的流动是反应性的,而外壳中的外部流动可以是反应性或非反应性的。通道壁是非渗透性的,只能通过它们进行热传递。
图7.2:通道及其周围外壳的横截面。
在进行Ansys Fluent迭代时,反应通道内的流动使用来自三维外部流动的固定壁温进行求解,然后使用来自通道求解器的固定热流求解三维外部流动。
7.1.5.2.1 反应通道内的流动
假设通道内的流动为活塞流,这意味着在任何轴向位置,流动变量在径向上是均匀的。轴向的热量和组分扩散也被忽略,因此仅通过反应发生变化。通道组分和温度的活塞流演化方程为
哪里,
混合物密度
整体温度
比热容
对流换热源
反应通道对流换热源 的确定如下
何处,
通道壁面温度 (请注意,不要与通道主体温度 混淆)是从解析的通道壁面上的三维外部流体温度场平均得出的,具体描述如下。
如第273页方程7.135所示,传热系数 的计算方法为:
其中, 和 分别代表气相热导率和通道直径。上述方程中的努塞尔数 是根据完全发展管流的实验相关性计算得出的,具体如下:
- 对于层流(雷诺数 Re )
对于湍流(雷诺数Re > 3000),采用Gnielinski关于湍流的关联式:
其中, 是摩擦系数。对于圆形截面的光滑管道, 的值由以下公式给出:
插值流动方程采用刚性ODE求解器求解,时间步长基于网格尺寸(通道元件的大小)和局部通道速度。
7.1.5.2.2 反应通道中的表面反应
当启用表面反应选项时,由于表面反应产生的额外源项被添加到方程7.134(第272页)中。关于表面反应引起的热量和质量源的详细信息,请参见壁面表面反应和化学气相沉积(第256页)。反应通道元件的反应表面积计算如下:
其中, 表示通道单元的体积, 是催化表面的面积与体积比。
注意:
在反应通道模型中,表面反应产生的热源以及质量沉积引起的热量和质量源始终被包含。
7.1.5.2.3 反应通道内的多孔介质
反应通道模型中的多孔介质选项采用表观速度多孔介质公式。Ansys Fluent 根据多孔区域内的体积流量计算表观混合速度。
当启用多孔介质选项时,从公式 7.136(第 273 页)计算得到的热传递系数会根据多孔介质中的固体材料进行如下修正。
其中, 表示孔隙率, 表示固体材料的导电性。此外,反应速率 通过乘以介质孔隙率 进行修正,以考虑气体被固体取代的影响。
Porous Medium 选项提供了模拟反应通道内压力下降的能力。通道中的压力下降仅在轴向方向上计算,并受到粘性阻力和惯性阻力的影响。
根据达西定律计算因粘性阻力引起的压降。
其中,表示沿轴向的粘性阻力,表示通道元件的长度。
由于惯性损失引起的压降被建模为
其中, 表示沿轴向的惯性阻力, 表示通道单元的长度。
7.1.5.2.4 壳体外流
外壳可以是任意形状的 3D 几何体,但必须解析通道壁。请注意,尽管在 Ansys Fluent 中使用有限体积单元对外壳进行网格划分,但通道内部不应进行网格划分。这种外流可以是反应性或非反应性混合物,通常与用于反应通道插管流的复杂机制材料不同。
Ansys Fluent 对外壳流进行能量求解时,使用从反应通道求解中得到的通道壁上的规定热通量边界条件。该热通量的值计算为从通道获得的热量的欠松弛:
其中, 表示通道的热量增益或损失, 是用户指定的欠松弛参数,而 则是上一迭代步的热通量。当收敛时,通道的热量损失或增益等于壳外流体的热量增益或损失。
7.1.6 Reactor Network模型
反应器网络模型用于使用详细的化学动力学机理模拟燃烧室中的组分和温度场。反应器网络由收敛的Ansys Fluent模拟构建,该模拟采用快速化学燃烧模型,如非预混、部分预混或涡耗散模型。可以将CHEMKIN格式的完整化学机理导入Fluent并在反应器网络中求解。燃烧室体积自动细分为少量连接的完全搅拌反应器。通过CFD解决方案确定网络中的质量通量,并在反应器网络中同时求解组分和温度。因此,反应器网络模型用于模拟有限速率化学效应与详细的动力学机理,特别是污染物排放如NOx、CO和未燃碳氢化合物。由于指定的搅拌反应器数量远少于CFD单元数量,与在每个单元中求解详细化学相比(如层流、EDC和PDF传输模型),反应器网络模型允许更快地模拟组分和温度场。
通常情况下,反应器网络模型在收敛的稳态RANS解决方案或时间平均的不稳定解决方案上执行。该模型也可以在不稳定的流动上运行,代表某个时间点的“快照”。由于没有将反应器-网络解决方案反向耦合到流动中,该模型对于预测详细化学对流动影响较小的情景非常有用,例如污染物形成。该模型不适用于高度不稳定的流动(如火焰点火或全局熄灭)以及受化学强烈影响的流动(如有显著烟灰辐射相互作用的烟灰)。
有关使用反应器网络的更多信息,请参阅Fluent用户指南中的Reactor Network Model部分
7.1.6.1 反应器网络模型理论
反应器网络模拟的第一步是将CFD单元格聚合成指定数量的反应器,。
由于每个反应器是燃烧室某个区域的完全混合表示,理想情况下,成分空间中最接近的单元格应分组在一起。为了优化性能,每个反应器中分组的CFD单元格应具有尽可能相似的温度和组分质量分数。
默认情况下,对于非预混和部分预混情况,Fluent将具有相似温度和混合分数的单元格分组;对于组分传输情况,Fluent将具有相似温度和及质量分数的单元格分组。这些默认设置在大多数情况下应提供良好的单元格聚类。然而,当这些默认设置不足时,Fluent允许通过自定义场函数进行用户控制的聚类。
在类似单元格被聚类后,Fluent拆分非连续组群,然后将包含最少数量单元的集群与其最近的邻居合并,直到达到指定数量的反应器为止。
反应器网络模拟的第二步涉及解决反应器网络问题,具体如下:
从CFD解中的单元格通量计算质量通量矩阵,其中每个矩阵组件是从反应器到反应器的质量通量。第种物质在反应器中的质量分数由以下代数方程控制:
where is the volume of reactor is the species reaction rate in the reactor, and is a mass source term. is the net mass flux into reactor and is calculated as:
质量源项 考虑了通过 CFD 边界进入的组分质量通量以及体积源(如离散相模型 DPM)的贡献。
方程组 ,即方程 7.146(第276页),其维度为 ,其中 是用户指定的反应器数量, 是化学机理中的组分数量。Ansys Fluent 默认使用分离算法求解此系统,但也可以选择使用完全耦合算法。
7.1.6.1.1 反应网络温度求解
在反应器网络中不解决能量方程。相反,默认情况下,每个反应器的温度通过状态方程计算。反应器压力是固定的,并确定为反应器中CFD单元的质量平均压力。请注意,由于反应器体积和质量都是恒定的,因此每个反应器的密度也是固定的。这种方法确保了在CFD模拟中的热损失(或增益)在反应器网络中得到适当考虑。Fluent还提供了一个选项,不计算温度,在这种情况下,温度固定为反应器中CFD单元的质量平均温度。