复杂化学
在 Simcenter STAR-CCM+ 中,使用刚性 CVODE ODE(常微分方程)求解器求解详细的复杂化学,从而对化学源项进行积分。考虑到湍流对燃烧的影响,还可以选择层流火焰概念 (LFC) 模型或涡耗散概念 (EDC) 模型。
复杂化学模型适用于将详细的化学信息引入 CFD 模拟。此模型可求解数百个组分中的数千个反应,因此称为复杂化学。由于使用 ODE 求解器对化学源项进行积分,因此复杂化学模型可以处理刚性反应系统(具有各种不同反应时间尺度的反应系统)。
在每个计算网格单元中应用于对化学源项进行积分的模型为恒压反应器。
反应系统
复杂化学模型需要有关组分、反应、热力学和传输属性的详细反应机制信息。这些详细信息由使用复杂化学模型以 Chemkin 格式导入的复杂化学定义文件提供。
源项定义
常规组分传输方程可设定为如下:
其中, 为扩散通量组分,且源项 为组分 的生成率。
此算子分裂算法将利用化学反应和流场涉及的不同时间尺度。化学状态(组分质量分数 和温度 )的时间积分将分两步执行:
- 在每个 CFD 网格单元中,每个时间步开始时,化学状态将从状态
到
进行积分,且仅用于化学源项:(3411)其中, 为刚性 ODE 求解器时间积分结束时的质量分数 (如同使用 Eqn. (3411) 进行计算)。 为Eqn. (3358)中的反应率, 为质量分数矢量, 为温度。
使用刚性 CVODE 求解器对 Eqn. (3411) 方程组进行求解。
- 组分传输方程将使用下式给出的第
个组分的显式反应源项
进行求解: (3412)其中, 为密度, 为平均反应率乘数, 表示单元网格中的当前质量分数。时间积分 指定用于非稳态模拟,并作为稳态模拟的停留时间 ,如Eqn. (3328)中定义。
对于非稳态模拟,必须保持低库朗数,以确保算子分裂格式中的任何误差均为较小值。
层流火焰概念 (LFC) 和涡耗散概念 (EDC) 通过提高的湍流扩散率(由湍流模型提供)隐式考虑湍流对燃烧的影响。
对于预混火焰,此提高的扩散率将导致火焰厚度大于层流火焰厚度,且湍流火焰传播速度快于层流火焰速度。火焰厚度和速度通过湍流施密特数和普朗特数(将这些数设为彼此相等)来控制。
除了湍流扩散率的这种隐式湍流效应之外,EDC 模型还考虑组分化学源中的“湍流化学相互作用”。对于 LFC 和 EDC,组分传输方程中的平均组分源项建模为Eqn. (3412)。
- 对于 LFC 模型,平均反应率乘数 (在 Eqn. (3412) 中)为 1,时间尺度 (在 Eqn. (3412) 中)为单元格中的停留时间,计算方法为 Eqn. (3328)。
-
其中, 为精细结构长度常数(默认值为 2.1377), 为运动粘度, 为湍流时间尺度, 为湍流长度尺度。
其中, 为湍流时间尺度,计算方法为一个常数(默认值为 0.4082)乘以 Kolmogorov 湍流时间尺度(定义为 )。
Eqn. (3411) 和 Eqn. (3412) 方程表示网格单元中的组分在时间步 中的平均变化率。LFC 时间步约为网格单元中的停留时间 Eqn. (3328)。Eqn. (3414)中的 EDC 时间步接近 Kolmogorov 时间尺度 (如相间湍流传输一节中所述),后者是最小湍流涡的时间尺度模型。由于 Kolmogorov 时间通常小于停留时间,且 Eqn. (3413) 中的 EDC 平均反应率乘数 小于 1,因此与 LFC 相比,EDC 通常将湍流的效应建模为平均反应率的减小。
- 化学平衡的松弛法假设化学成分按照由流体和化学时间尺度确定的时间尺度松弛到局部平衡成分。可以选择使用 ISAT 计算平衡成分。
化学源项根据指定的燃料组分计算得出:
(3415)其中, 为组分 的质量分数, 为组分 的局部瞬时热力学平衡质量分数, 为特征时间比例:
- 使用湍流火焰速度封闭 (TFC) 模型以外的模型时:(3416)
其中:
-
,其中,
为湍流耗散率,
为湍动能,
为湍流速率常数,默认值为 4。
对于层流 ,其中 为网格尺寸, 为局部速度。
- ,其中 是默认值为 2 的模型常数, 是用户指定的燃料组分列表的索引, 取所有用户指定燃料组分的最小值, 是 CFD 网格单元中的燃料组分质量分数,而 是组分 的净反应率。
-
,其中,
为湍流耗散率,
为湍动能,
为湍流速率常数,默认值为 4。
- 使用 TFC 模型时:(3417)其中 为时间尺度常数, 为 Kolmogorov 湍流时间尺度,如 所定义。
- 使用湍流火焰速度封闭 (TFC) 模型以外的模型时:
求解步骤
在 Simcenter STAR-CCM+ 中,可结合使用 CVODE 求解器与算子分裂算法求解反应组分输运方程,从而找到消除刚度的平均反应率。使用稳态模型运行时,将引入人工化学时间步,该时间步基于该网格单元中的对流和扩散通量。
在稳态模拟中,每个网格单元 的化学积分时间步 由下式确定:
这是网格单元体积 、密度 和中心系数 (由离散化和线性化得出)的函数。Eqn. (3418) 大约为网格单元中的停留时间 Eqn. (3328)。
复杂化学模型使用 CVODE 求解器对某个时间步的刚性化学进行积分。
数值雅可比表示使用有限差分法针对系统中的每个组分衍生得出的雅可比。由于化学源项 随 CVODE 求解器迭代而重复计算,因此求解 ODE 的成本十分高昂。
根据链式法则计算分析雅可比可获得 的分析表达式。
由于此过程的计算成本较高,因此使用 ISAT、聚类或动力机制减少以降低成本并加速达到收敛求解。
- ISAT
- 原位自适应制表 (ISAT) 以表格形式动态列出计算成本较高的函数,并在制表后多次进行插值[766]。ISAT 方法通过以下各项为给定的初始条件提供 ODE 近似求解 (Eqn. (3411)):
- 热力学变量(温度或总焓)。
- 成分变量(质量分数或摩尔数)。
- 可能包括压力和时间步(取决于模拟类型)。
- 聚类
- 聚类对具有相似化学成分的网格单元求平均值,并整合减少的 ODE 集合,然后将集群再次插值到网格单元中,以此减少计算复杂化学的计算开销。
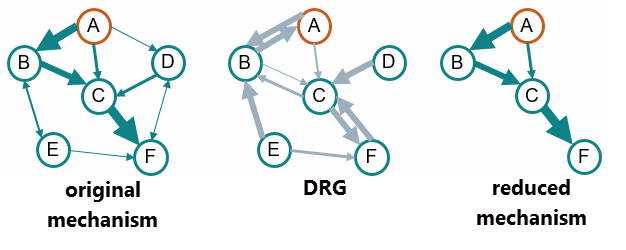
- 动态机理简化
- 通过动力机制减少,Simcenter STAR-CCM+ 中的 CVODE 求解器可以求解完整化学机制中减少的组分数量。定向关系图 (DRG) 算法 [755] 考虑每个时间步或每次迭代的网格单元中所有组分的反应率,如果组分在时间步中对任何其他组分的生成/破坏影响甚微,则会从机制中移除这些组分。
-